Product Planning
“Traditional” new drug development and approval—generally required for a new chemical entity drug that has not been approved before or that doesn’t have a significant marketing history in the U.S. or elsewhere—takes place under the provisions of section 505(b)(1) of the Federal Food, Drug, and Cosmetic Act. When it comes to 505(b)(1), the passage from “promising molecule” to “approved drug” is long, difficult, risky and expensive. Typically, achieving drug approval under the 505(b)(1) pathway—which requires the completion of new studies to establish the safety and efficacy of the drug in a specified disease or condition—can cost the sponsor up to 15 years and a billion dollars. Because a 505(b)(2) product can rely in part on the FDA’s previous findings on the safety and efficacy of an active ingredient as well as data available in the public domain, at this product planning stage, a potential developer of a 505(b)(2) should seek ways to weave such existing data into the product’s development strategy to reduce its size, scope, timeline and, therefore, cost.
In addition to representing a faster, less expensive path to market, products approved under the 505(b)(2) pathway can also sometimes qualify for several types of market exclusivity such as orphan drug exclusivity (seven years), new chemical entity exclusivity (five years), “other” exclusivity (three years for a “change” if certain criteria are met), and pediatric exclusivity (six months added to existing patents/exclusivity).
The accelerated path to approval and prospect of exclusivity make 505(b)(2) a cost-effective and commercially attractive route, but one with key differences from traditional 505(b)(1) development.
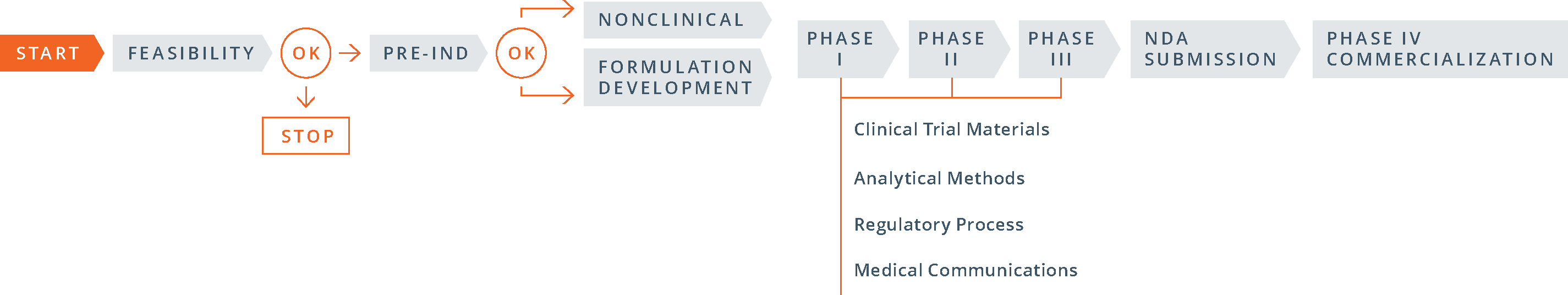
Pre-IND:
The 505(b)(1) pre-IND development process is fairly straightforward:
- conduct required nonclinical (animal) pharmacology, pharmacokinetics and toxicology studies; carry out early pre-formulation studies and select a lead formulation to advance; develop appropriate analytical methods; gather stability data on the active ingredient and the dosage form; and develop a proposed clinical protocol;
- complete a pre-IND consultation with the FDA in which the sponsor presents findings from its nonclinical studies and manufacturing and analytical data, as well as a proposed clinical trial, in order to gain FDA agreement to move to human testing; and
- file the investigational new drug (IND) application.
Compared to 505(b)(1), the 505(b)(2) process differs greatly. Here’s how:
- The order of the steps: The 505(b)(2) process begins with the pre-IND meeting with the FDA, then moves to formulation development (and studies, if necessary) and then to the IND filing.
- The goals of the pre-IND meeting: for a 505(b)(2) product PIND strategy is different than for a 505(b)(1). In proposing a 505(b)(2) development strategy in a pre-IND meeting, the objective is to gain FDA input and concurrence with the studies, with the chemistry, manufacturing, and controls (CMC) strategy and with clinical research plans in a way that minimizes the number of new studies required. For many companies, obtaining FDA buy-in and successfully activating an IND are critical steps for securing investments.
- The number and type of studies required: Since the 505(b)(2) pathway allows the use of public data or the FDA’s previous findings in lieu of novel trial data, some development programs may conduct bridging studies that preclude the need for nonclinical or clinical studies, or both.
- Timing of CMC work: For a 505(b)(2) product, the clinical trial materials for Phase I studies (often demonstrations of clinical bioequivalence) must be representative of the commercial manufacturing process, including packaging. In general, the three stability batches that will be used for shelf-life determinations are also prepared at this time. As a consequence, a good deal of CMC work must be invested prior to initiating even Phase I studies.
- Timing of studies: Because 505(b)(2) development plans rely largely on pre-existing data, and clinical studies can often be started simultaneously and developed in parallel, significantly shortening the overall time to market.
Together, these differences represent a formidable multifaceted challenge. When mishandled, the early steps of 505(b)(2) development can end in product-development failure. On the other hand, when managed skillfully, these first steps can result in important victories for the sponsor, including reduced costs, a clear path to approval and immediate interest from investors.
505(B)(2) BLOG ARTICLES
How to Achieve Success for 505(b)(2)