Clinical Trials With Multiple Endpoints: Pitfalls and Management
Randomized controlled Phase 3 (and some Phase 2) studies are clinical trials that are designed to answer specific questions, such as whether the proposed drug is effective in treatment or prevention of a particular disease. As such, the primary endpoint is the most important one that will demonstrate the primary effects being sought by the Sponsor. Subsequently, the sample size that is required to adequately power the study is dependent on the number of the events associated with the primary endpoint.
However, in many cases during development, the Sponsor may need clinical trials that are designed with multiple endpoints to demonstrate the efficacy and/or safety of a drug or biological product. Simultaneously, the statistical considerations during study design with respect to each of the endpoints need to be modified accordingly to reflect the influence of an endpoint to one another. If the design of a study with multiple endpoints does not appropriately take into account the effects of multiplicity, the study will not have the sufficient statistical power to support the claims, and results from an underpowered study can become a waste.
To this end, on 12 January 2017, the US Food and Drug Administration (FDA) announced the availability of the draft guidance entitled ‘‘Multiple Endpoints in Clinical Trials” (Docket No. FDA-2016-D-4460) for human drug/biologic assessments. This guidance provides helpful instructions for selecting appropriate endpoints and associated statistical methods to estimate a sizable clinical study with a sufficient power for clinical trials with multiple endpoints (CDER, 2017). Among the various statistical approaches presented in the guidance, each has pros and cons depending on the number and correlation levels of the endpoints (CDER, 2017).
Clinical Considerations
Efficacy endpoints are clinical events that vary depending on the intended effects of a drug on a particular disease. The clinical endpoints may be clinical events, patient symptoms, measures of function, or a family of events (such as scores) depending on the disease (e.g., Parkinson’s disease) or particular symptoms in a subset of patients (e.g., psychosis associated with Parkinson’s disease). In some cases, such as when the occurrence of a single event is low, efficacy can be based on a combination of several events (e.g., serum creatinine, end-stage renal disease, or death) that constitute a “composite event endpoint,” where a single event of the set would be analyzed as an “endpoint event.” In some other cases, multiple primary endpoints are needed to demonstrate the effectiveness of the investigational products (i.e., in Alzheimer’s disease, measure of cognition in combination with clinician’s global assessment or activities of daily living assessment would be co-primary endpoints) (CDER, 2017).
Statistical Considerations
There is a consensus that for a clinical trial with a single endpoint, the probability of finding a difference between treatment groups is set at 0.05 (α = 0.05). However, if there are two independent endpoints, both need to be demonstrated and both will be tested at α = 0.05, the chance of erroneously accepting successful conclusions (Type I error) is about 10 percent (1 – 0.95 × 0.95 = 0.0975). For three independent endpoints, the Type I error rate increases to 14 percent. Post hoc analyses cannot be used to demonstrate efficacy in trials intended to serve as the basis for FDA approval. In addition, endpoints that have been added after the completion of trials will not be accepted. Likewise, co-endpoints that were not adjusted for multiplicity when required will not be accepted, even if success can be demonstrated (CDER, 2017).
The Type I error (α), consumer’s risk, is the probability of concluding that the drug is effective when, in fact, there was none (a false-positive conclusion). The type II error (β), Sponsor’s risk, is the probability of concluding that the drug is not effective when in fact it is (a false-negative conclusion).
The likelihood of avoiding the Sponsor’s risk is called study power (1-β). The Sponsor would want to have a sizable study that provides sufficient power while demonstrating acceptable consumer’s risk. The use of co-primary endpoints (an effect on each is required for regulatory approval) will increase β, which leads to a decrease in statistical power. Further, in some cases, increasing the sample size may not be able to salvage study power in cases where co-primary endpoints are not fully independent. In cases in which – in addition to the primary endpoint family – Sponsors wish to have the study to be powered for one or two more secondary endpoints, estimation of an appropriate study sample size is difficult and may require virtual clinical trial simulation.
For this piece, Premier Consulting created a simulated example. Figure 1 below graphically depicts a case where one can reject the null hypothesis (a < 0.05), indicating that there is moderate evidence to support the alternative hypothesis (i.e., the drug is efficacious for the chosen clinical endpoint). However, the low power of the test (power = 1-β = 0.58) led to the failure of the study, likely due to the inadequate sample size estimation for the chosen primary endpoints and/or a poor choice of an effect size that is clinically meaningful.
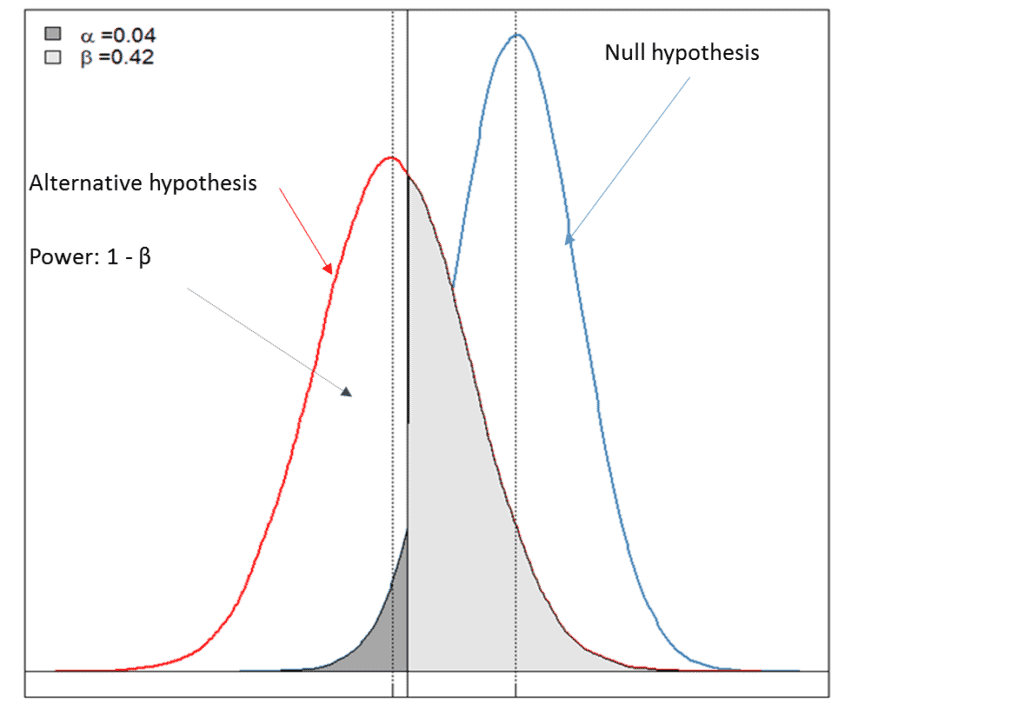
• Null hypothesis: there is no treatment effect on the chosen clinical endpoint of the test drug.
• Alternative hypothesis: there is at least some treatment effect on the chosen clinical endpoint of the test drug.
• α < 0.05: reject the null hypothesis and there is evidence to support the alternative hypothesis.
• Power of the test = 1-β and is dependent on the choice of effect size that is clinically meaningful and the sample size estimated for primary endpoints.
Conclusions
The likelihood of making a false-positive conclusion that a drug has a beneficial effect when it does not is the primary concern of the FDA. However, minimizing the Sponsor’s risk is equally important.
With deep expertise in drug development, Premier Consulting can provide consulting services for the identification and selection of clinical endpoints, suitable strategies and statistical methods at the study-planning stage to ensure the clinical trial will be appropriately designed and sufficiently powered so that the major findings are well supported by evidence obtained from the trials. Contact us to learn more about how we can help ensure the most time-efficient and cost-effective development path to NDA approval.
References:
CDER, CBER, Multiple Endpoints in Clinical Trials. Guidance for Industry. Draft Guidance. January 2017. Available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM536750.pdf