Advancing from Research to Development: What Can Go Wrong?
The drug development process is a long journey, beginning with drug discovery, moving through nonclinical and clinical studies, and ultimately culminating in regulatory approval. With many steps in between, each as important as the next, multiple factors regarding development strategy and approach must be considered at the earliest stages. This blog post offers advice for avoiding major pitfalls.
What can go wrong? Take a look.
Overview of a drug development program
The drug development process can be broken down into three main disciplines: chemistry, manufacturing, and control (CMC); nonclinical; and clinical. CMC programs include drug substance manufacturing process scale-up, stability testing, early formulation development, and, later on, manufacturing process validation, among other considerations. Nonclinical programs include (but are not limited to) method development and validation, pharmacokinetic studies, in vivo efficacy studies, safety pharmacology, IND-enabling toxicology studies, and long-term studies, when appropriate. A clinical program consists of the three main phases that evaluate the safety and efficacy of a drug in human subjects and patients.
Overview of drug development programs (through IND submission)
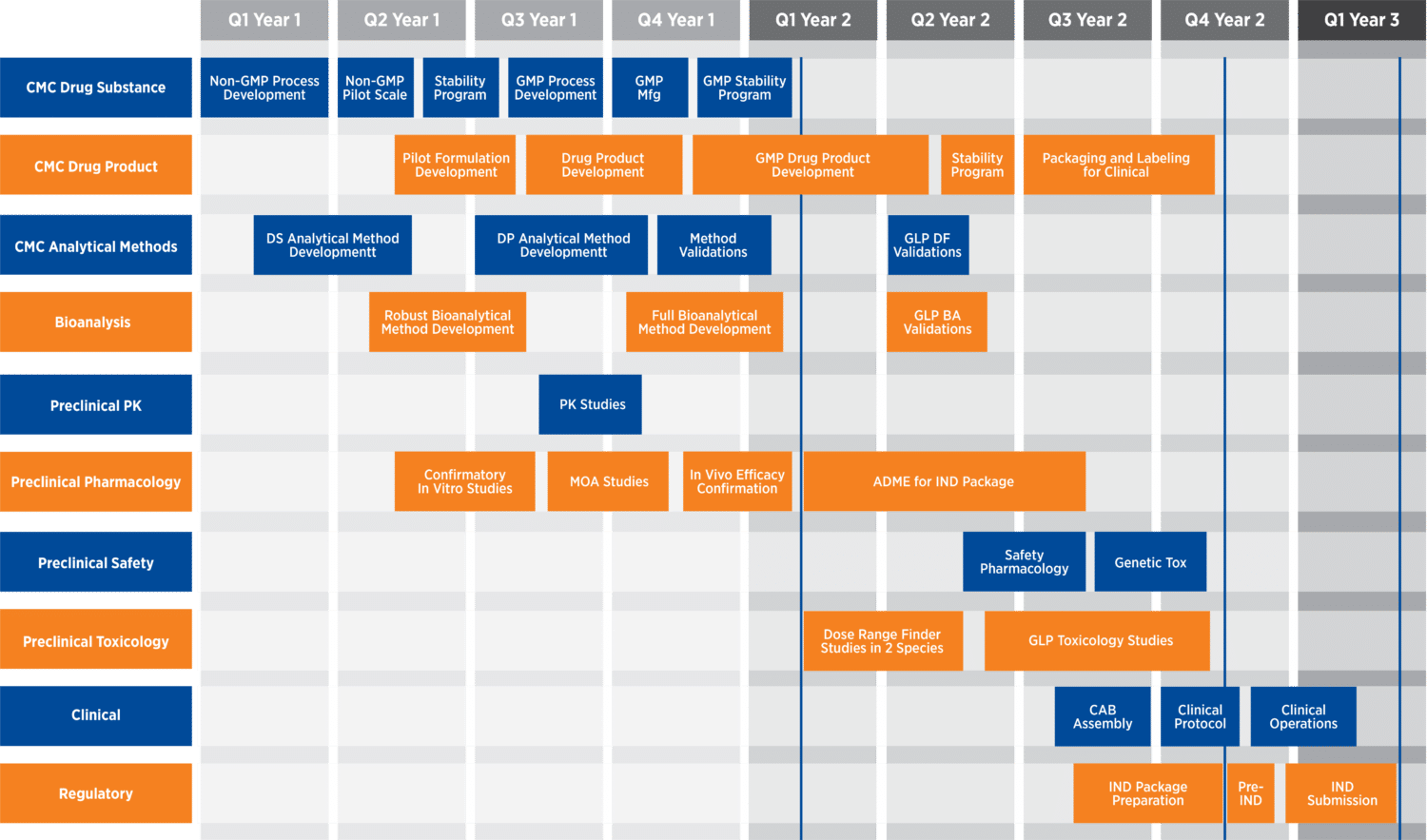
CMC considerations
Drug product formulation should be as simple as possible, and carefully executed to account for the characteristics of the intended administration route and dosing regimen and to facilitate successful manufacturing, storage, shipping, and compliance in the clinic and eventually on the market.
A drug substance ideally should maintain its characteristics throughout the development process. To optimize a CMC program, it is important to avoid certain issues:
- Too many steps in the manufacturing process
- Expensive starting materials
- Non-scalable processes
- Low yields
- High impurity or tedious purification steps
- Unstable intermediates
- Use of explosive reaction steps
The drug substance manufacturing process must be scalable to the commercial product while maintaining the characteristics of the drug. Manufacturing campaigns should be carefully planned to provide enough drug substance to execute all of the development activities, including stability studies, nonclinical and clinical studies, and supporting formulation development studies. These factors are important to ensuring that there is sufficient drug to execute each development stage and to ensure readiness for later-phase registration studies and regulatory approval.
Nonclinical considerations
Nonclinical studies are not mere tick-the-box exercises. A drug’s toxicology profile has a major influence on the approval process, and the FDA can issue a clinical hold and send a program back for further nonclinical studies. If this occurs, sponsors should not be discouraged — it is part of the process and happens with some frequency. However, it is preventable with proper analysis and planning of the toxicology program.
Early strategic planning
Toxicology programs must be defined early on. A strategic plan following a detailed gap analysis is needed to articulate all the necessary studies and their timing. When developing a strategy for a nonclinical program, sponsors should keep in mind the intended indication and the clinical studies to be conducted between the initial IND submission and the NDA submission.
GLP-compliant vs. non-GLP studies
It is important to bear in mind the differences between GLP-compliant and non-GLP studies when planning a nonclinical program. For example, in a GLP setting, validated analytical and bioanalytical methods must be in place, and dose formulation analysis must be included in the study plans. These standards minimize potential surprises, such as different matrix effects, stability issues, and longer-than-anticipated timelines.
Consistency across studies
Pharmacology studies must be aligned with a program’s toxicology program, and all studies in an overall development program must be conducted with the same administrative route and a similar vehicle. Intravenous delivery may not produce the same effect as inhalation, and the two should not be compared, as each route of administration has its own specific set of protocols.
Moreover, any changes to a program once studies are initiated must be carefully thought out, and changes to the API or dose formulation should be performed early in the program and avoided in later phases when possible. While formulation changes for an ongoing program may improve stability, they might also impact the bioavailability, toxicokinetic profile, or toxicology — potentially rendering all previous work void and irrelevant and costing the program money and time.
Species selection
The rodent and non-rodent species selected for toxicology studies should be based not only on scientific knowledge about the pharmacology, toxicity, and PK profile of a drug, but also on key early absorption, distribution, metabolism, and excretion data, if available. For example, sponsors should perform early metabolite profiling to ensure that all major human metabolites are represented in the selected species, rather than choosing based on initial study price or personal preference.
Study design
It is also important to avoid poor study design choices. For example, trying to save money by reducing the number of animals or dosing only one sex can be detrimental in certain study contexts. Here are several key recommendations for nonclinical study design:
- Do not overload a study with pharmacodynamic endpoints
- Avoid excessive bleeds, as they can affect blood chemistry and alter data
- Be sure that the doses administered are sufficient to allow measurement of observable dose-dependent toxic effects
Nonclinical team
Lastly, the sponsor should build a team ahead of time, with an appropriately experienced project manager and study director, and any other external expertise required for the program’s specific domain. It is also important to establish relationships among all members of the team so that all can focus on the same goal. Open communication, including monitoring and reporting, among all groups is essential to understanding and achieving program goals.
Clinical considerations
When designing clinical protocols, the patient profile should be considered carefully. Highly restrictive eligibility criteria could be scientifically sound but make recruitment impossible.
Functional areas should be discussed with a CRO from the beginning of study preparation to ensure proper organization of all study-related activities. Keeping study goals and endpoints in mind is crucial in this process. There are also several documentation considerations that should be established prior to start-up or early in the study to ensure smooth study conduct and regulatory and ethics compliance.
Among these considerations:
- Case report format and design
- Data management and biostatistical analysis plans
- Project management plan
- Dose escalating plan, if applicable
- Safety and medical monitoring plans
- External recruitment support expectations
- Trial master file plan
- Data transfer plan, if applicable
- Data monitoring plan
- Other study documents
Reviewing these suggestions prior to and during your drug development program can help ensure that it stays on track and help you sidestep pitfalls along the journey. The experts at Premier Partners and Premier Consulting have extensive experience navigating and solving these and other common issues in the transition from the research bench to drug development. Contact us today to find out how we can advance your program.
Author:
Alain Guimond, PhD
Vice President of R&D Operations
Premier Partners