505(b)(2) Approval Times: The Real Scoop
The Approval Time for 505(b)(2) and 505(b)(1) NME Products Is Similar
A recent article by the Tufts Center for the Study of Drug Development (summarized here) reported that approval times for New Molecular Entities (NMEs) approved via the 505(b)(2) pathway are nearly 5 months longer than that of NMEs approved by other pathways, 505(b)(1), or ‘traditional’ NDAs. Unfortunately, this misleading conclusion was based on a small number of approvals that included one outlier that required 5 review cycles and 94 months to approval. Removing this outlier would have led to the appropriate conclusion that the approval time for 505(b)(2) NME products is in fact similar to those of 505(b)(1) NMEs.
That review times are similar for products approved via 505(b)(1) and 505(b)(2) pathways is well known, as both pathways are subject to the same rigorous review processes and PDUFA commitments. The real time and cost saving for 505(b)(2) products comes from reduced development programs due to smaller and/or fewer studies rather than abbreviated review times.
Here, we use Premier Consulting’s proprietary 505(b)(2) database to look into the most common reasons for delayed approval times and we report that Chemistry, Manufacturing, and Controls (CMC) deficiencies top the list. Time and time again, we see that a thorough understanding of the 505(b)(2) pathway could have prevented costly delays in approval times.
505(b)(2) Products Approved in One Review Cycle
Of the applications approved via the 505(b)(2) regulatory pathway from 2009 – 2015, 64.5% were approved with only 1 review cycle. In some of these cases, preventable review delays occurred due to Refuse-To-File determinations, and the need to submit major amendments.
Common Reasons for Additional Review Cycles
Of the remaining applications in which more than 1 review cycle was undertaken, a staggering 73.2% involved CMC deficiencies. In 33.0% of applications, CMC deficiencies were the only factor in triggering a second or subsequent review cycle/s. It is a common misconception that CMC requirements are reduced for 505(b)(2) products compared with 505(b)(1). Stay tuned for our follow up blog for greater detail on CMC requirements.
Other common reasons for additional review cycles were issues with 505(b)(2) strategy (37.1%), and with demonstrating bioequivalence/conducting comparative bioavailability studies/scientific ‘bridging’ to other products (21.6%). These fundamental concepts in 505(b)(2) development programs are unique from 505(b)(1) programs and require experience to get right the first time.
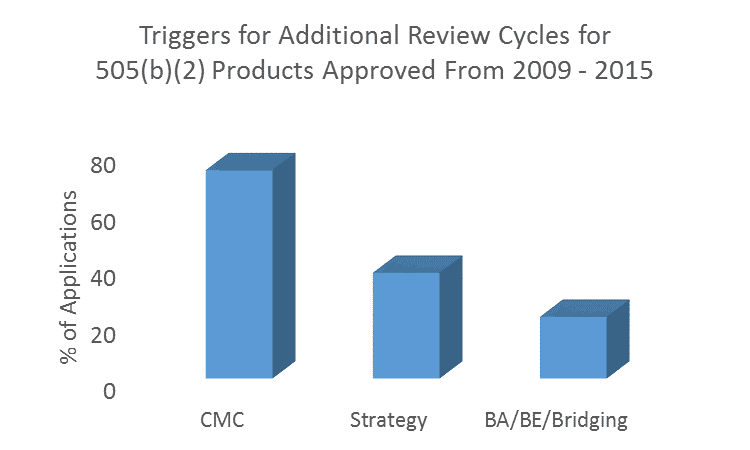
Other reasons for delays within the Sponsor’s control included formal dispute resolutions (4.1%), refuse-to-file determinations (3.0%), and data integrity issues (Application Integrity Policy, 2.1%). Delays that were largely out of the Sponsor’s control included changing standards (regulatory and/or clinical) including the need for advisory committee recommendations (9.3%), tentative approvals pending patent expiration (3.1%), or citizen petitions filed by Sponsors with competing products (1.0%). However, such delays rarely resulted in a new review cycle. Note that these numbers add up to more than 100% as some products were delayed for multiple reasons.
Outliers
Within the data, several outlier 505(b)(2) applications had much longer approval times. For instance, a product that was approvable from a clinical efficacy and safety perspective in the first review cycle was found to have major CMC deficiencies that dragged the program out for 4 additional review cycles with a resulting approval time of almost 8 years! Further, this product failed to obtain orphan exclusivity upon approval, as another product had beaten it to the punch by the approval date. Such costly mistakes could have been avoided with CMC oversight from individuals experienced in 505(b)(2) programs.
Within the period analyzed in the article (2009 – 2015), the longest ‘review time’ was just under 12 years. For most of the first 6 years of the 12-year review time for this single product, the review was suspended, as the application was placed under Application Integrity Policy for issues relating to data integrity.
In both of these examples and for many other products, demonstrating the clinical safety and efficacy of the proposed products was not a factor in the delayed approval time.
Effect of Priority Review on Approval
The Tufts analysis found that priority review had no beneficial effect on approval time for 505(b)(2) products. Unfortunately this is true for Sponsors that do not get their 505(b)(2) strategy right early in the development program. However, 9 of 33 (27.2%) products that were granted priority review were approved within 6 months via the 505(b)(2) pathway. In each of the remaining cases, the approval delay was due to deficiencies in the application. We cannot emphasize enough the importance of submitting a complete application based on sound strategy and with all required studies and appropriate pharmaceutical quality (CMC) data, especially if a Sponsor is hoping to enjoy the benefits of priority review.
Misconceptions on 505(b)(2) Approvals
As mentioned above, we note that with a thorough understanding of how the 505(b)(2) regulatory pathway works, shorter review/approval times are not expected for 505(b)(2) programs. The real advantage of the 505(b)(2) regulatory pathway is in the reduced scope of the development program. This translates into savings in time and cost for the Sponsor. But the review standards for 505(b)(2) programs are as rigorous as those of a 505(b)(1) NDA.
Secondly, the Tufts analysis compares products approved by the 505(b)(2) regulatory pathway with that of NMEs. However, it is important to understand what a 505(b)(2) is and isn’t in such a comparison, as a 505(b)(2) product can also be an NME. Further, some non-NME products are approved via the 505(b)(1) rather than the 505(b)(2) pathway. This helps to explain why the authors believe that “505(b)(2) review time goals mandated in PDUFA V for 505(b)(2) applications are two months shorter than for NMEs.” In fact, the PDUFA V goals distinguish between review times for NMEs (within 10 months of the 60-day filing review) and non-NMEs (within 10 months of receipt) under standard review (or within 6 months of the 60-day filing review vs. 6 months of receipt for NMEs and non-NMEs under priority review). So although many non-NMEs are 505(b)(2)s, it is an overstatement to suggest that 505(b)(2)s per se should be subject to shorter review times.
The same lack of understanding of the 505(b)(2) approval pathway causes the authors to state that “505(b)(2) applications rely heavily on data for previously approved drugs.” The authors cite this as a reason to expect shorter approval times for 505(b)(2) products. However, some approvals via the 505(b)(2) pathway do not rely on previously approved drugs. Some rely solely on reports in the literature. This explains why any difference in filing/review times is applicable to NMEs vs non-NMEs rather than 505(b)(2) vs NMEs.
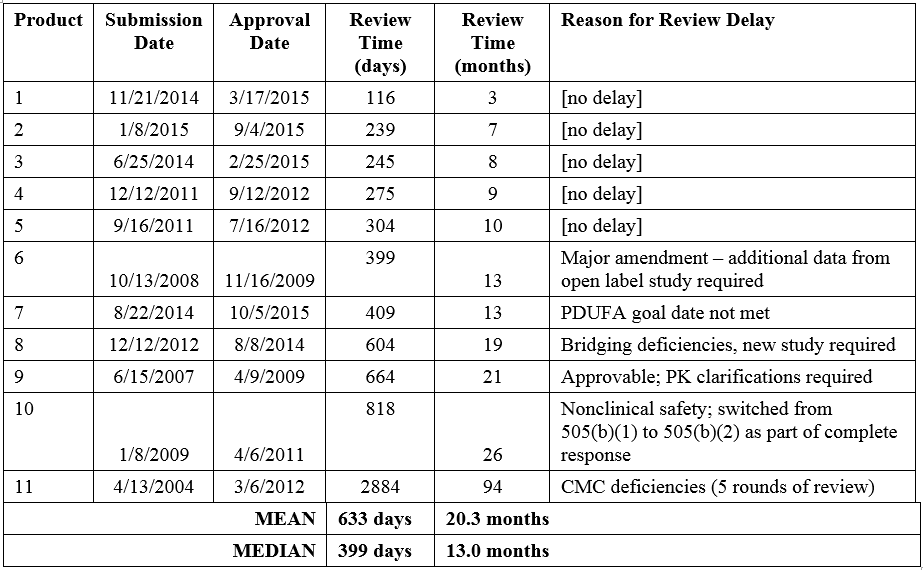
The Tufts analysis noted that the mean review time for the eleven NMEs submitted via the 505(b)(2) regulatory pathway was 55% longer than that of non-505(b)(2) NME approvals. The distribution of review times for the eleven 505(b)(2) NMEs ranges from 3 to 94 months, with a mean of 20.3 months (median 13.0 months; Table 1). The product with the longest review/approval time took more than 5 years longer than the next longest review time (26 months, Product 11). As discussed above, this product was approvable in the first review cycle with the exception of CMC deficiencies that took 4 more review cycles to resolve. When this outlier is excluded, the mean review time for 505(b)(2) NMEs drops to 12.9 months, similar to that of non-505(b)(2) NMEs (13.8 months).
It is also worth mentioning that the article defined approval time as the time from submission of the NDA to approval of the product. However, this includes Sponsor response time to FDA actions, and time for which review was suspended. For many of the applications with the longest approval times, Sponsor response times were significant.
Further, the authors did not specify if the duration between tentative approval and final approval was included in their calculation of approval time. It is worth noting that most formal review is complete at the time of tentative approval and that the remaining time is attributable to patent expiration issues with the listed drug rather than delays in FDA review or Sponsor responses. Tentative approval affects the final approval time of some 505(b)(2) and generic (505(j)) products, but not 505(b)(1) products.
Summary
There are many reasons why products approved via the 505(b)(2) pathway experience more than 1 review cycle and lengthy delays between cycles. Most of these reasons relate to CMC, development strategy, and pharmacokinetic studies. Almost all of these delays were preventable with proper oversight of the development program by individuals with expertise in 505(b)(2) approvals. As we recently blogged, it is critical to get 505(b)(2) strategy right the first time, particularly at the Pre-IND meeting, to save on time and costs. Premier Consulting’s 505(b)(2) experts have broad experience in CMC, strategy, gap analyses, and pharmacokinetic studies resulting in reduced approval time for Sponsors.
To talk to us about strategy for your 505(b)(2) product or how to minimize review cycles, read more here or contact us.
Author:
Angela Drew, PhD
Product Ideation Consultant